Panagiotis Alexiou, Manolis Maragkakis, Artemis G. Hatzigeorgiou
Biomedical Science Research Center Alexander Fleming, Vari, Greece
Correspondence: Panagiotis Alexiou, B.S.R.C. Alexander Fleming 34 Fleming Street, 16672, Vari, Greece.
E-mail: pan.alexiou@fleming.gr
Key words: microRNA, bioinformatics, online resources, computational, small RNA.
Conflict of interest: the authors report no conflicts of interest.
Received for publication: 15 November 2010.
Revision received: 14 February 2011.
Accepted for publication: 14 February 2011.
©Copyright P. Alexiou et al., 2011
Licensee PAGEPress, Italy
Journal of Nucleic Acids Investigation 2011; 2:e4
doi:10.4081/jnai.2011.e4
AbstractThe use of online tools for bioinformatics analyses is becoming increasingly widespread. Resources specific to the field of microRNAs are available, varying in scope and usability. Online tools are the most useful for casual as well as power users since they need no installation, are hardware independent and are used mostly through graphic user interfaces and links to external sources. Here, we present an overview of useful online resources that have to do with microRNA genomics, gene finding, target prediction and functional analysis. |
microRNAs are post-transcriptional regulatory molecules which belong in a recently identified group of short, 20-25 nucleotides long sequences of single-stranded, non-coding RNAs. microRNAs are produced by longer RNA precursors (pre-microRNA) whose length reaches approximately 100 nt. These precursors usually form an imperfect stem-loop structure (hairpin) and are in turn derived from longer primary RNA transcripts which can be thousands of nucleotides long and can contain several hairpins in transcriptional clusters.1
Generally, microRNA functionality derives from their base pairing on expressed mRNA molecules, usually on the 3’UTR but also on the coding sequence. This pairing, for animal microRNA in contrast to plant microRNAs, is rarely complete along the full length of the microRNA and can lead to degradation of the corresponding mRNA or to its translational repression.2
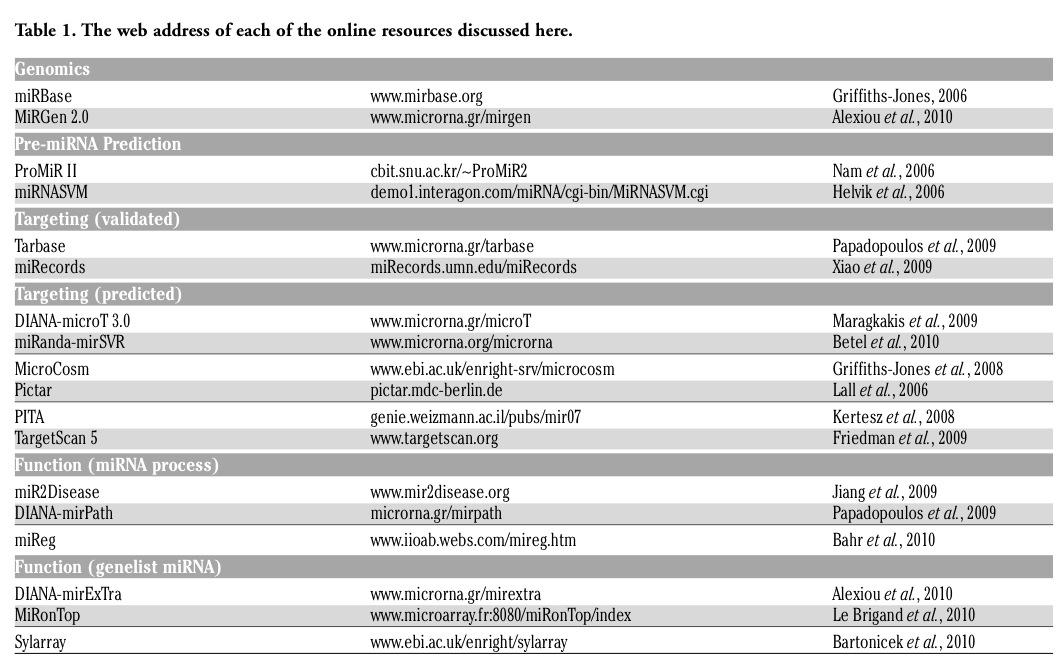
The discovery of microRNAs in the early 90s3 and their subsequent connection with a wide array of developmental programs and disease, has come in a time when bioinformatic techniques are becoming widespread, and the web all pervasive. Resources used by microRNA researchers on the web are numerous and continuously in flux. Here we present some of the most commonly used online resources in four categories sorted in alphabetical order per category (Figure 1, Table 1).
|
Figure 1. Online resources for microRNA analysis can be roughly divided in four categories. Genomic resources have to do with the genomic location and transcriptional interplay of microRNA genes. microRNA gene resources predict the hairpin structures associated with microRNAs. Targeting resources store experimentally validated or computationally predicted targets. Function resources show association of microRNAs with disease or function in general and of experimental results with microRNA deregulation. |
|
This category contains resources concerning genomic locations of microRNA primary transcripts, microRNA transcriptional clusters and genomic features associated with microRNAs such as transcription start sites and transcription factor binding sites near microRNA transcripts.
MiRBase 16.04 is a repository where newly discovered microRNAs are deposited and unique identification numbers are given. The basic unit of the database is the microRNA hairpin, with genomic location, sequence, references provided for hairpins in several species. The location and sequence of mature microRNAs on each hairpin is also provided. The database is searchable via an online interface, or can be downloaded as flat files and accessed offline.
miRGen 2.05 is a database that provides information on the genomic position and nearby features of human and mouse microRNA transcripts and cotranscribed microRNA clusters. Experimentally predicted transcription start sites and nearby predicted transcription factor binding sites are provided. Additionally, expression profiles of microRNAs in several tissues and cell lines, single nucleotide polymorphism locations, microRNA target prediction on protein coding genes and mapping of microRNA targets of co-regulated microRNAs on biological pathways are also integrated into the database and user interface.
The identification of novel microRNA genes,6 generally starts from the discovery of the distinctive hairpin structures that pre-microRNAs produce. With the onset of high-throughput experimental methods for the discovery of microRNA genes, the rate of identification of putative hairpin structures is ever increasing (Figure 2). There is a variety of online and offline tools for the prediction of the location of pre-microRNA hairpins in given sequences or genomic locations.
Among the on-line tools: miRNASVM7 is a machine learning classifier that predicts the processing sites for Drosha, the Class 2 RNase III enzyme that processes pre-microRNAs. The classifier attempts to find 5′ Drosha processing sites in hairpins that are candidate microRNAs thus attempting to separate true from false microRNA hairpin predictions.
ProMiR II8 is a web-server that identifies microRNA hairpin structures in given sequences. It consists of three distinct programs. One searches for novel microRNA hairpins near known microRNAs, one predicts hairpins near a candidate sequence, and the last one is more general, using a moving window approach to scan larger sequences. Several parameters and thresholds can be set by the user.
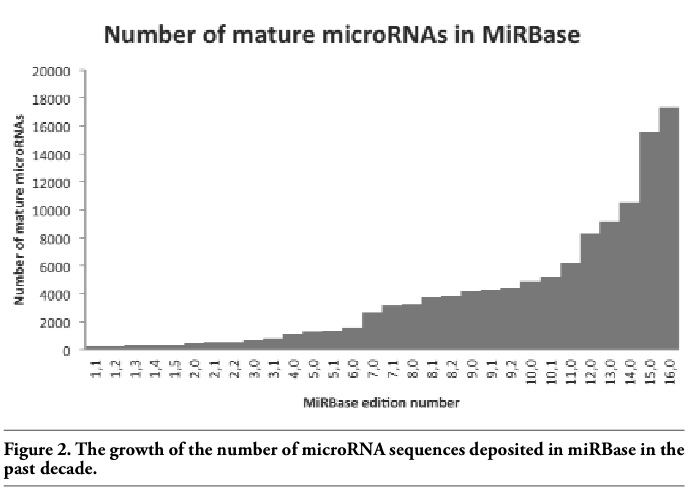 |
Figure 2. The growth of the number of microRNA sequences deposited in miRBase in the past decade. |
Experimental validation of microRNA targets has been progressing in bounds in the past few years. Besides the more direct methods of target validation employing luciferase constructs and other traditional molecular biology methods, a great increase in high-throughput validation methods has been evident in the past few years.
miRecords9 is an integrated resource for animal microRNA-target interactions. The Validated Targets component of this resource hosts a manually curated database of experimentally validated microRNA-target interactions with systematic documentation of experimental support for each interaction. The current release of this database includes 1135 records of validated microRNA-target interactions between 301 microRNAs and 902 target genes in seven animal species. The Predicted Targets component of miRecords stores predicted microRNA targets produced by 11 established microRNA target prediction programs.
Tarbase10 is a database which houses a manually curated collection of experimentally supported microRNA targets in several species. The current version includes more than 1300 experimentally supported targets. Each target site is described by the microRNA that binds it, the gene in which it occurs, the nature of the experiments that were conducted to test it and other factors. The whole database can be accessed online or downloaded.
Although it is becoming increasingly easier to experimentally validate microRNA targets of interest, the computational prediction of microRNA targets is still relevant. The use of novel high-throughput experimental methods allows researchers to obtain a wider range of known microRNA targets. Such data will probably help in the identification of new rules that govern microRNA function and also serve as training sets for applications based on machine learning approaches. As expression data is becoming increasingly available, it will be soon possible to train adaptive algorithms that will highlight additional rules for miRNA interactions with targeted genes. However, to date, most microRNA target prediction programs are based on fixed rules. Since the field of microRNA target prediction is very fast changing and competitive with large differences in performance among programs. An overview of the performance of microRNA target prediction programs on high-throughput experimental data shows great discrepancies in the predictive strengths of each method.11 Here we provide a brief overview of the most accurate and widely used microRNA target prediction programs.
DIANA-microT 3.012 is an algorithm based on several parameters calculated individually for each microRNA and it combines conserved and non-conserved microRNA recognition elements into a final prediction score. The program reports a signal to noise ratio and a precision score which help in the evaluation of the significance of the predicted results. The web server provides extensive information for predicted microRNA:target gene interactions providing extensive connectivity to online biological resources. Target gene and microRNA functions may be elucidated through automated bibliographic searches and functional information is accessible through KEGG pathways. The web server offers links to nomenclature, sequence and protein databases and users are facilitated by being able to search for targeted genes using different nomenclatures or functional features, such as the genes possible involvement in biological pathways.
MicroCosm13 (formely known as miRBase Targets) uses the miRanda algorithm to initially identify potential binding sites for a given microRNA. Dynamic programming alignment is used to identify highly complementary sites. Strict complementarity at the microRNA seed region is demanded. Thermodynamic stability is estimated for each target site. For inclusion in the database, conservation of the target site at the exact same position in at least two species is required.
miRanda - mirSVR14 mirSVR is a new machine learning method for ranking microRNA target sites by a down-regulation score. The algorithm trains a regression model on sequence and contextual features extracted from miRanda-predicted target sites. In a large-scale evaluation, miRanda-mirSVR is competitive with other target prediction methods in identifying target genes and predicting the extent of their downregulation at the mRNA or protein levels. Importantly, the method identifies a significant number of experimentally determined non-canonical and non-conserved sites.
PicTar15 identifies microRNA targets with perfect or imperfect complementarity in a 7nt seed region. Conservation is taken into account, and an HMM approach provides the final score by combining the multiple microRNA targets identified on the same gene. Although PicTar is still relatively accurate11 when compared to other microRNA prediction algorithms, it has not been updated to the latest identified microRNAs since its initial release, thus missing hundreds of newly identified microRNAs.
PITA16 incorporates binding site structural accessibility as a feature and does not take into account the evolutionary conservation of the binding site. Although, it is not among the best performing programs11,17 it remains an interesting approach that shows high potential of being used along with other prediction programs that are more dependent to the evolutionary conservation of binding sites.
TargetScan 5.118 is one of the most widely used microRNA target prediction programs. In TargetScan, microRNA binding sites are predicted through the identification of seed matches on the 3’UTR of mRNAs and the assessment of their evolutionary conservation across several species. The overall scoring of a microRNA binding site denoted as context score depends on binding features such as whether the identified match involves binding on position 8 and/or whether it has an A at position 1, the localization of the binding site within the 3’UTR and the AU content of the area flanking the binding site. The final prediction score indicating whether a microRNA target a particular gene is calculated by summing the context scores of all corresponding binding sites identified on that gene 3’UTR.
A field of interest for many researchers is the function of microRNAs. When a list of microRNAs or a list of known or putative microRNA targets is given, a researcher would be interested to find out whether they are associated with any diseases or physiological processes.
DIANA-mirPath19 is a web-based computational tool developed to identify molecular pathways potentially altered by the expression of single or multiple microRNAs. The software performs an enrichment analysis of multiple microRNA target genes comparing each set of microRNA targets to all known KEGG pathways. The combinatorial effect of co-expressed microRNAs in the modulation of a given pathway is taken into account by the simultaneous analysis of multiple microRNAs. The graphical output of the program provides an overview of the parts of the pathway modulated by microRNAs, facilitating the interpretation and presentation of the analysis results.
miR2Disease20 is a manually curated database which aims at providing a comprehensive resource of microRNA deregulation in various human diseases mined from published data. Users can also suggest associations based on publications.
miReg
miReg21 is a manually curated microRNA Regulation Resource that represents regulatory relationships between TFs, microRNAs and other regulators. The information is based on published resources.
Although microRNA expression levels may not be routinely measured in high-throughput experiments, a possible involvement of microRNAs in the deregulation of gene expression can be computationally predicted. Especially with the increasing use of high-throughput expression arrays and sequencing to measure deregulation in mRNA and even protein levels, these techniques for the computational prediction of the possible involvement of microRNAs are becoming more relevant.
DIANA-mirExTra22 allows the comparison of frequencies of microRNA associated motifs between sets of genes that can lead to the identification of microRNAs responsible for the deregulation of large numbers of genes.
MiRonTop23 is an online java web tool that integrates DNA microarrays or high-throughput sequencing data to identify the potential implication of microRNAs on a specific biological system. It also provides useful representations of the enrichment scores according to the position of the target site along the 3’-UTR, where the contribution of the sites located in the vicinity of the stop codon and of the polyA tail can be clearly highlighted. It provides different graphs of microRNA enrichment associated with up- or down-regulated transcripts and different summary tables about selections of mRNA targets and their functional annotations by Gene Ontology.
SylArray24 is a web-based analysis resource designed to examine influence of small RNAs on expression profiles. It can be used to find significant enrichment or depletion of microRNA or siRNA seed sequences from microarray expression data.
As an increasing number of resources in the fields related to microRNAs are becoming available it is of the greatest importance for users to know which resources are available in order to be able to choose which one to use for a specific task. The onset of the sequencing era in genomics brings great expectations for all the sub-fields of microRNA analysis. Databases will need to scale accordingly to increasing data loads and user requests, machine learning approaches will be used more often and in wider scopes and possibly user generated content could start being used. In closing, we would like to apologize to the large number of groups that work in this field whose work was impossible to be included in this review.
1. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281-97. [Pubmed]
2. Filipowicz W, Jaskiewicz L, Kolb FA, Pillai RS. Post-transcriptional gene silencing by siRNAs and miRNAs. Curr Opin Struct Biol 2005;15:331-41. [Pubmed]
3. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993;75:843-54. [Pubmed]
4. Griffiths-Jones S. miRBase: the microRNA sequence database. Methods Mol Biol 2006;342:129-38. [Pubmed]
5. Alexiou P, Vergoulis T, Gleditzsch M, et al. miRGen 2.0: a database of microRNA genomic information and regulation. Nucleic Acids Res 2010;38:D137-41. [Pubmed]
6. Oulas A, Reczko M, Poirazi P. MicroRNAs and Cancer inverted question mark The Search Begins! IEEE Trans Inf Technol Biomed 2008 Aug 15. [Pubmed]
7. Helvik SA, Snove O, Jr, Saetrom P. Reliable prediction of Drosha processing sites improves microRNA gene prediction. Bioinformatics 2007;23:142-9. [Pubmed]
8. Nam JW, Kim J, Kim SK, Zhang BT. ProMiR II: a web server for the probabilistic prediction of clustered, nonclustered, conserved and nonconserved microRNAs. Nucleic Acids Res 2006;34:W455-8. [Pubmed]
9. Xiao F, Zuo Z, Cai G, et al. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res 2009;37:D105-10. [Pubmed]
10. Papadopoulos GL, Reczko M, Simossis VA, et al. The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res 2009;37:D155-8. [Pubmed]
11. Alexiou P, Maragkakis M, Papadopoulos GL, et al. Lost in translation: an assessment and perspective for computational microRNA target identification. Bioinformatics 2009;25:3049-55. [Pubmed]
12. Maragkakis M, Alexiou P, Papadopoulos GL, et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinformatics 2009;10:295. [Pubmed]
13. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res 2008;36: D154-8. [Pubmed]
14. Betel D, Koppal A, Agius P, et al. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol 2010;11:R90. [Pubmed]
15. Lall S, Grun D, Krek A, et al. A genome-wide map of conserved microRNA targets in C. elegans. Curr Biol 2006;16:460-71. [Pubmed]
16. Kertesz M, Iovino N, Unnerstall U, et al. The role of site accessibility in microRNA target recognition. Nat Genet 2007;39:1278-84. [Pubmed]
17. Selbach M, Schwanhausser B, Thierfelder N, et al. Widespread changes in protein synthesis induced by microRNAs. Nature 2008;455:58-63. [Pubmed]
18. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009;19:92-105. [Pubmed]
19. Papadopoulos GL, Alexiou P, Maragkakis M, et al. DIANA-mirPath: Integrating human and mouse microRNAs in pathways. Bioinformatics 2009;25:1991-3. [Pubmed]
20. Jiang Q, Wang Y, Hao Y, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res 2009;37:D98-104. [Pubmed]
21. Barh D, Bhat D, Viero C. miReg: a resource for microRNA regulation. J Integr Bioinform 2010;7. [Pubmed]
22. Alexiou P, Maragkakis M, Papadopoulos GL, et al. The DIANA-mirExTra web server: from gene expression data to microRNA function. PLoS One 2010;5:e9171. [Pubmed]
23. Le Brigand K, Robbe-Sermesant K, Mari B, Barbry P. MiRonTop: mining microRNAs targets across large scale gene expression studies. Bioinformatics 2010;26:3131-2. [Pubmed]
24. Bartonicek N, Enright AJ. SylArray: a web server for automated detection of miRNA effects from expression data. Bioinformatics 2010;26:2900-1. [Pubmed]
[TOP]