Koshi Mimori, Takehiko Yokobori, Masaaki Iwatsuki, Tomoya Sudo, Fumiaki Tanaka, Kohei Shibata, Hideshi Ishii, Masaki Mori
Department of Surgical Oncology, Medical Institute of Bioregulation, Kyushu University, Beppu and Department of Gastroenterological Surgery, Graduate School of Medicine, Osaka University, Suita, Japan
Correspondence: Masaki Mori, Department of Gastroenterological Surgery, Graduate School of Medicine, Osaka University, Yamadaoka 2-2, Suita 565-0871, Japan. E-mail: mmori@gesurg.med.osaka-u.ac.jp
Key words: fragile histidine triad, esophageal cancer, microarray, apoptosis, inflammation, adenoviral transduction.
Acknowledgment: we express our appreciation to T. Shimooka, K. Ogata, M. Kasagi and T. Kawano for their technical assistance. This work was supported in part by the following grants and foundations: CREST, Japan Science and Technology Agency (JST); Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research, grant numbers 20012039, 20390360, 20590313, 20591547, 20659209, 20790960, 21591644, 21592014, 21791295, 21791297, 215921014, 21229015 and 21679006; NEDO (New Energy and Industrial Technology Development Organization) Technological Development for Chromosome Analysis; Grant of Clinical Research Foundation (2008-2010).
Received for publication: 30 October 2009.
Revision received: 6 May 2010.
Accepted for publication: 8 May 2010.
©Copyright K. Mimori et al., 2010
Licensee PAGEPress, Italy
Journal of Nucleic Acids Investigation 2010; 1:e7
doi:10.4081/jnai.2010.e7
AbstractWe focused on the mechanism by which FHIT suppresses neoplastic transformation in normal but damaged esophageal epithelial cells exposed to inflammatory stimuli in vivo and to chemo-radiotherapy in clinical samples.
|
Esophageal cancer, considered to be one of the most intractable human malignancies, can be initiated by several different environmental carcinogens. Pandeya et al. showed that alcohol intake significantly increases the risk of squamous cell carcinoma of the esophagus, and smoking modifies the effect of alcohol intake on risk.1 Continuous exposure to alcohol and smoke damages esophageal epithelial cells and accelerates malignant transformation. In addition to consumption of alcohol and exposure to myosmine in smoke, several inflammatory processes, such as gastro duodenal reflex disease, also gave rise to esophageal cancer.2,3,4,5 In addition, continuous inflammation of colorectal epithelium gives rise to malignant transformation. However, the presence of FHIT prevents inflammatory bowel disease-mediated cancer as we reported in our previous study.6,7 There are few studies of FHIT, a tumor suppressor molecule, and of the mechanism by which it suppresses the actions of inflammatory carcinogens in the esophagus.
In our previous study, we showed that fragile histidine triad (FHIT), which is located on 3p14.2 within a common fragile region, was diminished in most epithelial malignancies, including esophageal cancer, due to an alteration of genomic DNA.8,9,10 In addition, the FHIT gene was repressed in normal esophageal epithelium in healthy individuals who were exposed to large amounts of alcohol and tobacco smoke.11 We hypothesized that continuous inflammatory stimulation of epithelial cells might play a causative role in carcinogenic activity. Therefore, we examined the role of FHIT protein and the severe inflammatory cascade initiated by carcinogens. We found that inflammatory stimulation of FHIT-expressing esophageal cancer cell lines did not lead to activation of prostaglandin E2 (PGE2) (as evaluated by ELISA assay). Furthermore, cellular proliferation was reduced as measured by MTT assay.6 The inhibition of FHIT expression by FHIT siRNA demonstrated up-regulated expression of PGE2 accompanied by cellular proliferation as we hypothesized in our previous in vitro study.6 Supplementing our previous in vitro work, in the current study, we clarified the in vivo biological response of epithelial cells lacking FHIT to inflammatory carcinogenic stimulants in the esophagus and the forestomach of mice.
Then, we focused on how FHIT inactivated those carcinogenic signals and thereby suppressed tumorigenesis in the esophagus. For example, does FHIT respond as a mere anti-inflammatory agent, such as NSAID or steroid? We hypothesized that FHIT might induce apoptosis in damaged epithelial cells to avoid initiating a tumorigenic cascade due to excessive inflammatory stimuli. In this regard, we recently disclosed that FHIT protein is involved in surveillance of genome integrity and checkpoint response after genotoxin exposure.12 Tissues of genotoxin-exposed FHIT-deficient bone marrow-transplanted mice exhibited preneoplastic alterations, including accumulation of DNA damage. Those carcinogenic stimuli allowed long-term survival of genotoxin-exposed FHIT-deficient hematopoietic stem cells with deleterious mutations.
Therefore, in the current in vivo study, we show that FHIT-deficient esophageal epithelial cells with enhanced carcinogenic potential might develop into tumors due to the PGE2-mediated inflammatory cascade. In addition, to determine the role of FHIT in human esophageal cancer, we asked whether FHIT activated esophageal cancer can induce apoptosis following cellular insults, such as chemo-radiotherapy treatment.
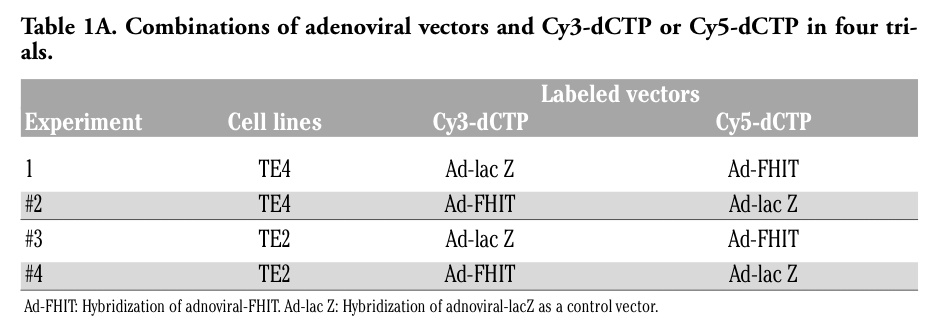
We previously reported the expression of genes following adenoviral attenuation of FHIT.13,14 In the current study, we established a gene expression profile, focusing on the PGE2 synthetic pathway following adenoviral-induced expression of FHIT. As shown in Table 1A, in order to improve the reliability of the expression profile of adenoviral-FHIT compared with adenoviral-LacZ vector, we performed 7 repetitive hybridizations among three cancer cell lines lacking endogenous FHIT expression: TE4 and TE2 (esophageal cancer, provided by Dr. Y Shimada, Department of Surgery and Basic Surgical Research, Kyoto University).14,15
Total RNAs were extracted and 2.5 µg aliquots of mRNA from either Ad-FHIT or control infected samples were labeled with Cy3-dCTP or Cy5-dCTP (Amersham, Biotech). Labeled probes were hybridized for 14-16 hr at 65°C with a chip, a total number of 7 times altogether. The chip was printed with 38384 oligos consisting of 19,192 genes prepared at the Microarray Facility of the Kimmel Cancer Center, Thomas Jefferson University. After hybridization, the slides were washed twice in 2x SSC/0.2% SDS for 30 min at 55°C and 2x SSC/0.2% SDS for 30 min at 65°C, then 0.05x SSC for 5 min at room temperature. Immediately, thereafter, the slides were scanned. The intensity of each hybridization signal was evaluated by a computer program and the normalization of such expression was performed by housekeeping genes on the chip. After hybridization and bio-informatics processing, the list of Ad-FHIT repressed genes in the TE4 and TE2 infected cells were obtained. As shown in Table 1A, the Ad-FHIT/control expression ratio was determined based on informative data points (all experiments were performed 2 or 3 times using duplicate spots per gene on each chip) for each gene. After the above repetitive in vitro experiments, we calculated the average expression ratio of Ad-FHIT/Ad-LacZ in TE4 and TE2 individually.
Eight to ten week old male mice with a wild-type or FHIT-deficient genotype in a C57BL/6 background were divided into four experimental groups: i) 7 wild-type mice were given N-nitrosomethylbenzylamine (NMBA);16 ii) 6 FHIT-deficient mice were given NMBA; iii) 7 wild-type mice were given both NMBA and cyclooxygenase-2 inhibitor, celecoxib (COXI);17 iv) 6 FHIT-deficient mice were given both NMBA and COXI. For the NMBA group, NMBA was administered by drinking water (6 mg/L) for eight weeks. For the NMBA plus COXI group, the administration of COXI (40 mg/L) was started four days before NMBA (6 mg/L), and the administration of both reagents continued for eight weeks. All mice were sacrificed to excise the forestomachs which were fixed in buffered formalin and prepared for hematoxylin and eosin (H&E) staining and immunohistochemistry. A part of the epithelium in each tissue was dissected from the remaining tissues (using a blade to strip off the connective tissue layer), snap frozen in liquid nitrogen and stored in -80°C for the PGE2 assay.
We performed immunohistochemical studies of 26 specimens from the forestomach epithelium from mice, and 10 representative lesions from 10 patients with esophageal cancer. 4 µm sections were prepared for tissue slides. We incubated slides with primary rabbit FHIT antiserum against the C terminus of the FHIT protein (1:1,000 dilution, overnight, Zymed), BAX (2772, Cell Signaling; N-20, Santa Cruz), COX2 and PCNA antisera, followed by incubation with appropriate biotinylated secondary antibodies.
Slides were then incubated with streptavidin horseradish peroxidase (Dako; 1:1,000 dilution). Sections of normal tissue were used as a reference control for staining and scoring. The intensity of expression of FHIT, BAX and PCNA was scored (as described previously) as follows: 0, no staining; 1+, less staining than normal epithelium; 2+, similar to normal epithelium; 3+, stronger than normal epithelium.
Results
In Table 1B, we analyzed up-regulated genes in the PGE2 synthetic pathway in apoptotic cells in which adenovirus attenuated FHIT. We found a statistically significant lower expression of cyclooxygenase-2 (COX-2) in adenoviral-FHIT cells than control cells (P=0.032). In addition, other molecules related to the synthesis of PGE2 were significantly reduced by up-regulated expression of FHIT in cancer cells, including COX-1 (P=0.006), early growth response-1 (P=0.022), and PGE receptor 4 (P=0.025). Therefore, FHIT should not inactivate PGE2 synthetic pathway genes directly and specifically; thus, FHIT might inactivate any biological activity in any cancer cells. Note also that adenoviral-FHIT transduction induced more apoptosis in TE4 cells than in TE2, and that expression of the above mentioned genes was much lower in apoptotic TE4 cells than in non-apoptotic cell TE2 cells (data not shown).
Among the four groups subjected to NMBA exposure (wild-type mice, wild-type mice with COXI, FHIT-deficient mice and FHIT-deficient mice with COXI), a significant difference was observed in PGE2 production between FHIT wild-type mice (5.2 ng/mL) and FHIT-deficient mice (28.4 ng/mL) without COXI by ELISA assay (P<0.01) (Figure 1). In other words, PGE2 was significantly more abundant in FHIT-deficient mice than in wild-type FHIT mice. On the other hand, the simultaneous administration of NMBA and COXI eliminated the significant difference in PGE2 level in the forestomach between animals with and without FHIT.
In the immunohistochemical analyses of the four groups, BAX expression in FHIT wild-type was significantly higher than that in FHIT-deficient mice (P<0.047). There was no significant difference in COX2 expression between FHIT wild-type mice and FHIT-deficient mice (Figure 1). PCNA expression was higher in FHIT-deficient mice than in FHIT wild-type mice in spite of the administration of COXI; however, there was no statistically significant difference between them.
Chemo-radiotherapy responsive lesions in esophageal cancer revealed expression of FHIT and BAX
Before comparing the expression of FHIT, BAX, and PCNA in esophageal tumors after chemo-radiotherapy, it was important to deal with the tumors’ variable responsiveness to therapy. Therefore, we hypothesized that it would be helpful to separate the responsive and non-responsive tumors into two groups. Thus, we compared expression of FHIT, BAX and PCNA between responsive lesions and non-responsive lesions in esophageal tumors after chemo-radiotherapy (Table 2). The IHC score of FHIT expression was 2.33±0.578 in the 3 responsive lesions, while there was no FHIT expression in the 7 non-responsive lesions (P<0.05). As for BAX expression, 3 responsive lesions scored significantly higher (2.67±0.58) than 7 non-responsive lesions (0.14±0.38) (P<0.05). PCNA expression in 7 non-responsive lesions was three times higher than in 3 responsive lesions; however, there was no statistically significant difference between them. Those findings are demonstrated in 4 cases in Figure 2. In cases #1 and 3, non-responsive lesions showed robust expression of PCNA; however, neither FHIT nor BAX was observed in these identical cases. On the other hand, in cases #2 and 9, chemo-radio therapy-responsive lesions with necrotic tissues showed FHIT and BAX expression, while PCNA expression was weak.
|
Table 1A. Combinations of adenoviral vectors and Cy3-dCTP or Cy5-dCTP in four trials. |
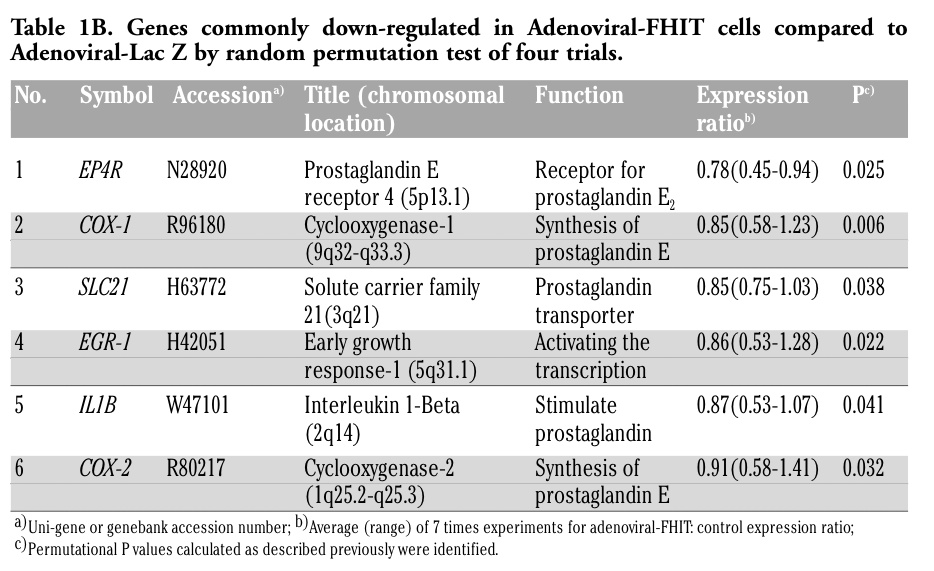 |
Table 1B. Genes commonly down-regulated in Adenoviral-FHIT cells compared to Adenoviral-Lac Z by random permutation test of four trials. |
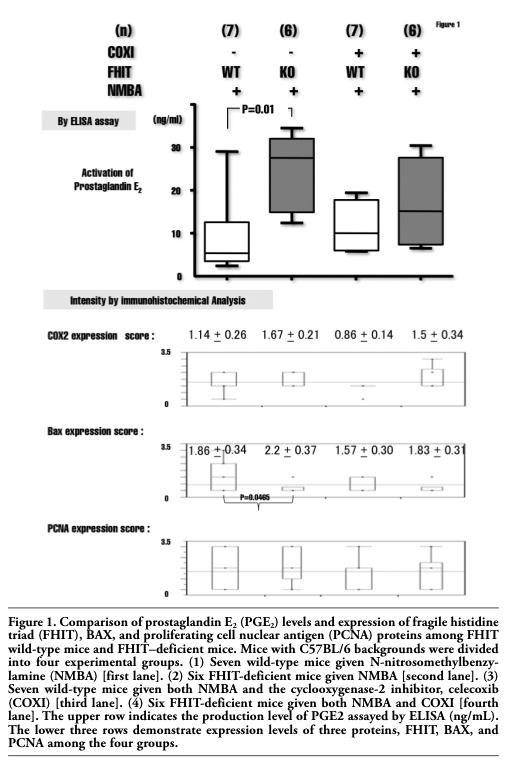 |
Table 2. Comparison of expression of FHIT, BAX and PCNA between responsive lesions and non-responsive lesions in esophageal tumors after chemoradiotherapy. |
|
Figure 1. Comparison of prostaglandin E2 (PGE2) levels and expression of fragile histidine triad (FHIT), BAX, and proliferating cell nuclear antigen (PCNA) proteins among FHIT wild-type mice and FHIT–deficient mice. Mice with C57BL/6 backgrounds were divided into four experimental groups. (1) Seven wild-type mice given N-nitrosomethylbenzylamine (NMBA) [first lane]. (2) Six FHIT-deficient mice given NMBA [second lane]. (3) Seven wild-type mice given both NMBA and the cyclooxygenase-2 inhibitor, celecoxib (COXI) [third lane]. (4) Six FHIT-deficient mice given both NMBA and COXI [fourth lane]. The upper row indicates the production level of PGE2 assayed by ELISA (ng/mL). The lower three rows demonstrate expression levels of three proteins, FHIT, BAX, and PCNA among the four groups. |
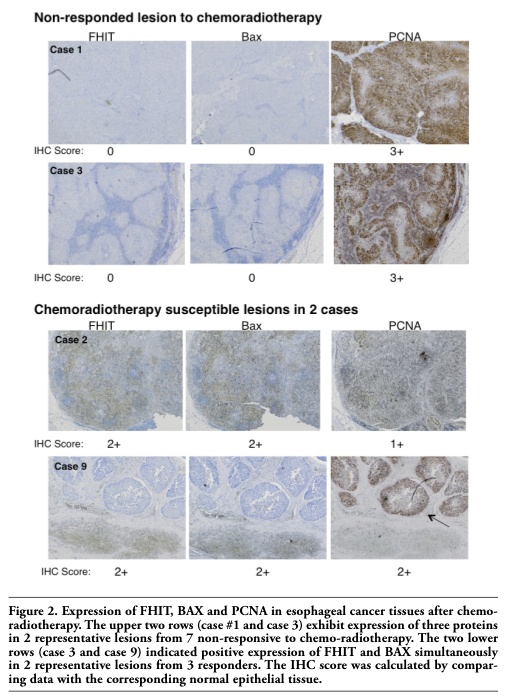 |
Figure 2. Expression of FHIT, BAX and PCNA in esophageal cancer tissues after chemo-radiotherapy. The upper two rows (case #1 and case 3) exhibit expression of three proteins in 2 representative lesions from 7 non-responsive to chemo-radiotherapy. The two lower rows (case 3 and case 9) indicated positive expression of FHIT and BAX simultaneously in 2 representative lesions from 3 responders. The IHC score was calculated by comparing data with the corresponding normal epithelial tissue. |
In the current study, we demonstrated that esophageal epithelial cells from FHIT-deficient mice increased their production of PGE2 and reduced the frequency of apoptosis in vivo following exposure to NMBA.18 Considering this evidence, we conclude that FHIT promotes apoptosis in normal but damaged epithelial cells by the inflammatory stimuli as well as in damaged carcinoma cells in the esophagus by the chemo-radiotherapy. After exposure to cellular damage, NMBA in vivo and chemo-radiotherapy during therapeutic treatment, we speculated that the number of surviving cells with the capacity for malignant transformation increases and therefore PGE2 production derived from those non-apoptotic malignant cells increases accordingly.
In our previous study, we reported a direct correlation between PGE2 synthesis and FHIT expression in colorectal cancer cells, suggesting that FHITs postulated tumor suppressive effect operates through PGE2 but not COX-2 according to our immunohistochemical data. We concluded that FHIT protein did not inhibit a specific molecule in the arachidonic acid cascade, but directly and specifically PGE2 activity.6 On the other hand in esophageal cancer according to the expressing genes’ profile in Adeno-FHIT attenuated cells rather than controls, we found downregulation of whole pro-inflammatory molecules in the arachidonic acid pathway, such as PGE2, COX-1, COX-2 and receptors for PGE2. We assumed that FHIT induces apoptosis in cells damaged by inflammatory carcinogens; therefore, total PGE2 production and synthetic pathway molecules are presumably reduced in FHIT-expressing apoptotic cells. In esophageal cancer, FHIT does not work as a mere anti-inflammatory molecule, but induces apoptosis buffering the cellular DNA damage by strong inflammatory stimuli leading to malignancies.
As for the chemo-radiotherapy damaged esophageal cancer tissues, FHIT induced apoptosis of damaged esophageal cancer cells as a response to the chemo-radiotherapy, regardless of absence of local inflammation in esophageal cancer tissues. In other words, the absence of FHIT protein in each case mostly due to the genetic alteration in 3p14 locus,9 a fragile site, might not be able to induce apoptosis nor response to the chemo-radiotherapy in cancer tissues regardless of the presence of inflammation.
In conclusion, we demonstrate here that FHIT might not directly affect specific molecules regulating the PGE2 synthetic pathway, but might instead inactivate whole damaged cells and thereby decrease the amount of metabolic products by inducing apoptosis after exposure to the DNA damaging inflammatory carcinogens and to DNA damaging chemo-radiotherapy.
1. Pandeya N, Williams G, Green AC, et al. Alcohol consumption and the risks of adenocarcinoma and squamous cell carcinoma of the esophagus. Gastroenterol 2009 136:1215-24.[PubMed]
2. Chen T, Stoner GD. Inducible nitric oxide synthase expression in N-nitrosomethylbenzylamine (NMBA)-induced rat esophageal tumorigenesis. Mol Carcinogen 2004;40:232-40.[PubMed]
3. Devlin AH, McIlroy M, McKeen HD, et al. Cytochrome P450 1B1 expression in rat esophageal tumorigenesis promoted by gastric and duodenal reflux. Mol Carcinogen 2009;48:110-7.[PubMed]
4. Fein M, Peters JH, DeMeester TR. Carcinogenesis in reflux disease--in search for bile-specific effects. Microsurgery 2007;27:647-50.[PubMed]
5. Vogt S, Fuchs K, Richter E. Genotoxic effects of myosmine in a human esophageal adenocarcinoma cell line. Toxicology 2006;222:71-9.[PubMed]
6. Mimori K, Ishii H, Nagahara H, et al. FHIT is up-regulated by inflammatory stimuli and inhibits prostaglandin E2-mediated cancer progression. Cancer Res 2006;66: 2683-90.[PubMed]
7. Xu CM, Qiao CH. Loss of fragile histidine triad protein expression in inflammatory bowel disease. World J Gastroenterol 2006;12:7355-60.[PubMed]
8. Inoue H, Ishii H, Alder H, et al. Sequence of the FRA3B common fragile region: implications for the mechanism of FHIT deletion. Proc Natl Acad Sci USA 1997; 94:14584-9.[PubMed]
9. Mimori K, Druck T, Inoue H, et al. Cancer-specific chromosome alterations in the constitutive fragile region FRA3B. Proc Natl Acad Sci USA 1999;96:7456-61.[PubMed]
10. Ohta M, Inoue H, Cotticelli MG, et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 1996;84: 587-97.[PubMed]
11. Mori M, Mimori K, Shiraishi T, et al. Altered expression of Fhit in carcinoma and precarcinomatous lesions of the esophagus. Cancer Res 2000;60:1177-82.[PubMed]
12. Ishii H, Mimori K, Ishikawa K, et al. Fhit-deficient hematopoietic stem cells survive hydroquinone exposure carrying precancerous changes. Cancer Res 2008;68:3662-70.[PubMed]
13. Ishii H, Dumon KR, Vecchione A, et al. Effect of adenoviral transduction of the fragile histidine triad gene into eso-phageal cancer cells. Cancer Res 2001; 61: 1578-84.[PubMed]
14. Mimori K, Ishii H, Inoue H, et al. Identification of the expression profile of apoptotic esophageal cancer cells by adenoviral-fragile histidine triad treatment. J Gastroenterol Hepatol 2008;23 Suppl 2:S205-9.[PubMed]
15. Shimada Y, Imamura M, Wagata T, et al. Characterization of 21 newly established esophageal cancer cell lines. Cancer 1992;69:277-84.[PubMed]
16. Wargovich MJ, Woods C, Eng VW, et al. Chemoprevention of N-nitrosomethylbenzylamine-induced esophageal cancer in rats by the naturally occurring thioether, diallyl sulfide. Cancer Res 1988;48:6872-5.[PubMed]
17. Reddy BS, Hirose Y, Lubet R, et al. Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res 2000;60:293-7.[PubMed]
18. Ishii H, Zanesi N, Vecchione A, et al. Regression of upper gastric cancer in mice by FHIT gene delivery. FASEB J 2003;17: 1768-70. [PubMed]
[TOP]